Computer simulations are useful for developing conceptual understanding of science ideas that are otherwise obscure. However, there are circumstances that simulations just raise more questions than what they answer. Osmosis is one of those deceptively simple phenomena that turn out to be quite challenging to understand, even if it can be simulated with reasonable clarity.
 |
| Figure 1. Osmotic pressure. (Image from Wikipedia) |
Osmosis is a process in which solvent molecules move -- without input of energy -- across a semipermeable membrane (which let only the solvent molecules pass) separating two solutions of different concentrations. A typical explanation is that the solvent molecules "want" to equalize the solute concentrations (or equivalently, the solvent concentrations) on the two sides. As a result, the solutions must build up persistent pressure that causes the liquid level in the left side of the U-shape tube shown in Figure 1 to rise remarkably higher against gravity.
Most people just walk away with this theory. But where exactly does the energy that elevates the liquid come from?
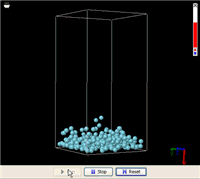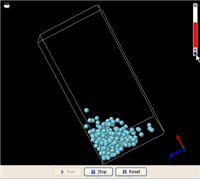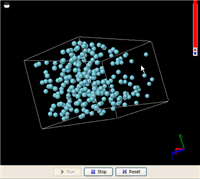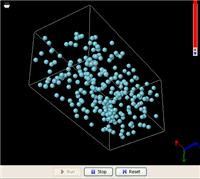
In the U-shape tube, no one exerts any force on the liquid in either side, while the force of gravity keeps the liquid level as low as possible. Somehow, by simply making the membrane in the middle partially permeable to only the solvent, some energy is extracted to do the heavy lifting. This process can be simulated using molecular dynamics as is shown in the YouTube video posted above. The simulation shows that, on average, the middle column eventually maintained a noticeably higher level. Removing the solute (the green particles) returned the liquid levels in the three columns to about the same.
In this simulation, the green particles and the blue particles have comparable chemical affinities, meaning that the blue particles do not particularly favor their kins around. Neither do the green particles. The white particles that represent the membrane molecules have very weak interactions with both blue and green particles.
Here is the link to the simulation (which is a Java applet). Happy New Year!